


前言
随着Liraglutide及Semaglutide在治疗T2D方面心血管事件的获益,无疑对GLP-1受体激动剂药物在未来糖尿病市场的竞争中增加了一枚很有分量的砝码。诺和诺德近期公布的Semaglutide在减肥适应症的数据,更是令人兴奋。随着口服GLP-1药物三期临床的大规模开展,以及GLP-1类药物独特的优势,越来越多的公司加入到了GLP-1药物开发的大军中。
GLP-1的发现
GLP-1最初发现于20世纪80年代早期,随后被证实可以通过胰岛β细胞以葡萄糖依赖的方式促进胰岛素的分泌。GLP-1的分泌源自于肠道内分泌细胞对膳食的应答,从而刺激餐后胰岛素的释放。早期,科学家以口服葡萄糖可以促进胰岛素分泌的现象为基础,进行GLP-1药物的开发。然而,GLP-1会被DPP-4酶迅速的裂解而失活,因此内源性的GLP-1无法开发为治疗药物。因此,DPP-4抑制剂可以通过增加激素稳定性,从而增加GLP-1的活性。此类化合物已经被证实,可以有效的控制血糖,特别是联合二甲双胍使用,并且同时表现出可以增强内源性GLP-1的释放。
GLP-1合成及作用机制
GLP-1合成之后,会被肠道内分泌细胞中的前胰高血糖素原基因加工。在α细胞中,前胰高血糖素原转化为胰高血糖素和主要的胰高血糖素原片段;而在肠道L细胞中,前体激素会被PC1/3酶转化为GLP-1,GLP-2,肠高血糖素和氧调蛋白。GLP-1与胰高血糖素同源50%,可以诱导葡萄糖刺激的胰岛β细胞释放胰岛素。GLP-1主要以两种活性形式存在,GLP-1(7-36氨基化合物)和GLP-1(7-37氨基化合物)。此多肽可以在DPP-4酶的作用下迅速失活,通过裂解N端的HIS-ALA二肽。
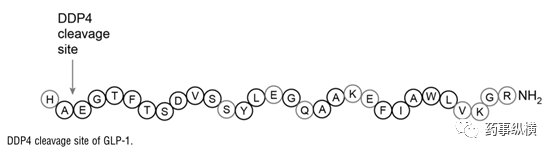
GLP-1R在多个组织中均有表达,包括:胰岛细胞、大脑以及肠道。GLP-1R是G蛋白偶联受体B家族的GPCR,有一个大的胞外区。活性受体在胰岛β细胞,可以增加cAMP,激活PKA,同时增加胰岛素的生物合成。在高血糖的情况下,可通过cAMP的积累促进β细胞释放胰岛素。正常情况下,以葡萄糖依赖的形式,激活GLP-1R刺激胰岛素的分泌。
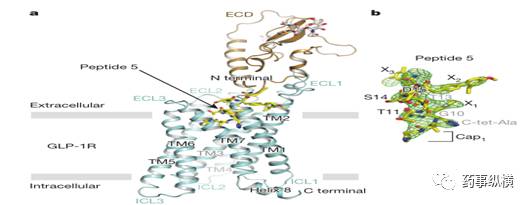
GLP-1构效关系
对GLP-1构效关系的研究发现,GLP-1的N端对于受体的激活是至关重要的,C端在与受体胞外区结合方面也很关键。
Pos1
HIS在GLP-1R结合与激活方面均扮演重要角色,在任何一个GLP-1类似物中均存在。通过HIS的修饰,可以抵抗蛋白酶的降解,例如:N甲基化、α甲基化、去氨基化以及咪唑乳酸均能抑制DPP-4酶的降解。但是,完全替换HIS会影响GLP-1的活性。在用ARG或者LYS取代时,会降低活性;用PHE取代活性的降低是最小的。
Pos2
N端第二个氨基酸ALA的修饰,被广泛的研究,在修饰后可抑制DPP-4酶对GLP-1的降解,延长其半衰期。在Liraglutide中,ALA就被GLY取代,只是轻微的降低了活性,而很大程度上增加了稳定性。但是SER的取代,会使与受体的结合活性降低10倍,空间结构中使得LEU和isoLEU被掩盖,因此是效能显著降低。由Aib取代效果最佳,不仅可以保留活性,而且达到了抑制DPP-4酶降解的目的。
Pos3
电负性的GLU和ASP在受体的结合与效能上是非常关键的,用碱性氨基酸取代会引起效能的显著降低。ALA、SER及VAL的取代也会使得效能下降,氨基酸手性的改变也会使效能大打折扣。
Pos4
4位的GLY在与GLP-1R相互作用中起到非常重要的角色。
Pos5-8
5-8位氨基酸的组合对于活性是非常关键的。
Pos9
电负性氨基酸ASP对活性是关键的,替换为GLU会轻微的影响亲和性。手性的改变,也会对亲和性有一定的影响。
Pos10-15
10位的VAL替换为疏水氨基酸会丢失一部分活性和亲和性;12位的SER被ALA取代不会受到影响,但是被LYS取代时亲和性会下降四倍。15位的GLU被小分子量的氨基酸取代,会显著降低亲和性;11、13及14位的氨基酸在与GLP-1R结合和活性方面也是重要的。
Pos16-20
16位的GLY可以被任何氨基酸取代,而不丢失活性;20位的LYS通常被用来修饰;17-19位的氨基酸对活性是关键的。
Pos21-25
21位的GLU被任何氨基酸取代均会极小的损失与GLP-1R的亲和性;21位被碱性氨基酸取代不会丢失活性;22位的PHE对于活性和亲和性都是至关重要的;23位的ILE在与受体结合方面是关键的;24和25位会对活性有影响。
Pos26-31
26位的LEU对于活性是重要的,被全氟丁二烯类似物取代在效能和亲和性方面会有轻微的影响;27位的VAL同样在天然和合成的GLP-1中都存在;28位的LYS是另一个很好的修饰位点;29位的GLY被Aib取代后,可以提供更好的半衰期;30及31位的氨基酸在受体结合和活性方面都是非常关键的。
首个GLP-1药物
DPP-4抑制剂开发的同时,更加稳定的内源性GLP-1类似物被发现。Exenatide,发现于毒蜥唾液中的激素exendin-4,可以通过人工合成的方式进行制备,为首个上市的GLP-1类似物药物。Exenatide较GLP-1序列更长,具有55%的同源性。更具优势的特点为,Exenatide将GLP-1中更易被DPP-4裂解的丙氨酸以甘氨酸进行了取代,从而增加了稳定性并延长了半衰期。

2位的GLY增加了稳定性,阻止DPP-4酶的降解。21及39位氨基酸形成一个紧密的结构,保护Trp25侧链在溶剂中尽量少的暴露,从而增加多肽和α螺旋的稳定性。
GLP-1上市药物
Lixienatide

Lixienatide包含的氨基酸残基在1-39位与Exenatide相似,只是少一个Pro,在C端增加了6个Lys。Lixienatide日剂量20µg,半衰期为3h。在联合二甲双胍质量糖尿病方面,低血糖的发生率要低于Exenatide。
Taspoglutide

Taspoglutide与天然的GLP-1非常接近,在2位与29位替换为了Aib。这样的修饰增加了稳定性,可以防止DPP-4酶、血浆酶的降解。三期临床的缓释制剂,对HbA1c有持续稳定的作用。最终由于免疫原性及过敏性,最终被终止。
Liraglutide
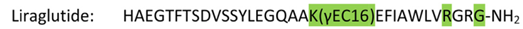
Liraglutide结构与天然GLP-1(7-37)有97%氨基酸残基是相似的。在28位由Arg取代了Lys,在20位的Lys通过谷氨酸酯链接了棕榈酸侧链。棕榈酸侧链的链接,增加了与血清白蛋白的结合,从而延长了半衰期,Liraglutide在人血浆半衰期为11-13h。不仅在质量T2D上有很好的效果,如果剂量增加至3mg,减重也可以达到6%以上。
Semaglutide

Semaglutide是由诺和诺德开发的长效GLP-1激动剂,对于T2D一周注射一次的治疗方案已经提交FDA及EMA的注册申请。与Liraglutide结构有两个差别,2位的Gly被Aib取代,脂肪酸侧链不同。这两点的不同,导致Semaglutide的半衰期延长至160h。并且,Semaglutide的口服制剂已进入三期临床阶段。
Albiglutide

Albiglutide是将两个GLP-1类似物以重组的方式,融合至人血清蛋白的N端。为防止DPP-4酶的降解,将2位的Ala替换为Gly。这样重复单元的串联方式,增加了与白蛋白的结合能力,半衰期为6-8天。
Dulaglutide

Dulaglutide由礼来公司开发,为GLP-1融合FC蛋白的结构。2位的Ala与30位的Arg被Gly替换,从而防止DPP-4酶的降解。16位的Glu提高了化合物的效能。半衰期约为4天。
结语
GLP-1类似物在T2D治疗方面,无低血糖副作用,并且有心血管获益,可预见在未来的T2D市场竞争中肯定会带来让人惊喜的表现。口服Semaglutide的开发,更是在口服胰岛素无法突破的情况下,将会是一枚重磅炸弹。减肥方面的优异表现,也给开发NASH适应症带来了很大的希望。因此,GLP-1受体激动剂药物一定会让大家欣喜。
